Short Running Head
JAKi in EGPA
Full Title of Manuscript
JAK inhibitors in Eosinophilic Granulomatosis with Polyangiitis (EGPA): an international case series
Authors
- Paul Anthony John Russo, ORCiD ID: 0000-0003-1190-7161
- Jean-Paul Makhzoum, ORCiD ID: 0000-0003-4523-8525
- Vincent Cottin, ORCiD ID: 0000-0002-5591-0955
- Edoardo Conticini, ORCiD ID: 0000-0002-3974-6606
- Giacomo Emmi, ORCiD ID: 0000-0001-9575-8321
- Samuel Lawrance Whittle, ORCiD ID: 0000-0001-7417-7691
Key Indexing Terms
Eosinophilic granulomatosis with polyangiitis, Janus kinase inhibitor (JAK inhibitor), treatment
Sources of support
Dr Paul Anthony John Russo would like to acknowledge the support of Arthritis Australia and The Australian Rheumatology Association Research Fund Private Practice Grant for 2025.
Author Affiliations
- Dr PAJ Russo B.Med.Sci (Hons), MBBS, FRACP, FACR: Private practice: Chandlers Hill Surgery, Adelaide, South Australia, Australia
- J-P Makhzoum MD. MSc. FRCPC, FACP: Associate Professor, Université de Montréal, Service de Médecine Interne, Hôpital du Sacré-Coeur de Montréal, Université de Montréal, Canada
- V Cottin MD, PhD: Université Claude Bernard Lyon 1, Hospices Civils de Lyon, Centre de référence des maladies pulmonaires rares, Service de pneumologie, ERN-LUNG, INRAE, UMR754, F-69000 Lyon, France
- E Conticini MD, PhD: Rheumatology Unit, Department of Medicine, Surgery and Neurosciences, University of Siena, Italy
- G Emmi MD, PhD: Professor of Internal Medicine, Department of Medical, Surgical and Health Sciences, University of Trieste, Trieste, Italy; Centre for Inflammatory Diseases, Monash University Department of Medicine, Monash Medical Centre, Melbourne, Australia
- SL Whittle MBBS(Hons) MClinEpi FRACP: Rheumatology Unit, Queen Elizabeth Hospital, Woodville, South Australia, Australia
Conflicts of Interest
Dr Paul Anthony John Russo has received lecturing and consulting fees from AbbVie, Johnson & Johnson and GSK. Dr Vincent Cottin has received lecturing fees from Astra Zeneca and GSK. Dr Giacomo Emmi has received lecturing fees from Astra Zeneca, GSK, Novartis, SOBI and AbbVie.
Corresponding Author
Dr Paul Anthony John Russo
Chandlers Hill Surgery
194A Chandlers Hill Road
Happy Valley 5159, South Australia
Australia
Email: pajrusso@drpaulrusso.com.au
Ethics
This study was approved by the Central Adelaide Local Health Network (CALHN) Human Research Ethics Committee and CALHN Research Services (reference number: 19079). All patients described in this paper provided informed, written consent.
Abstract
Objective
Despite significant recent advances in the management of eosinophilic granulomatosis with polyangiitis (EGPA), glucocorticoid toxicity remains a major issue. Janus kinase inhibitors (JAKi) provide a mechanistically sound theoretical option for the treatment of EGPA. Here, we report our collective experience of 5 patients with EGPA who received JAKi therapy.
Methods
Physicians around the globe with an interest in vasculitis were contacted in 2024 and early 2025 in order to identify patients with EGPA who had received treatment a JAKi (tofacitinib, baricitinib, upadacitinib or ruxolitinib).
Results
Five patients were identified, two from Australia (managed in private practice), one from Canada (tertiary referral centre), one from France (tertiary referral centre) and one from Italy (tertiary referral centre).
Conclusions
Despite the fact that only a small number of cases were identified, impressive clinical responses to JAKi therapy were observed, providing a strong argument for a formal randomized controlled trial comparing JAKi therapy to the current standard of care for EGPA.
Introduction
First reported by Churg and Strauss in 1951 [1], eosinophilic granulomatosis with polyangiitis (EGPA) is a rare small vessel vasculitis histologically characterized by eosinophil-predominant granulomatous inflammation, tissue eosinophilia and necrotizing vasculitis. Anti-neutrophil cytoplasm antibodies (ANCA) are detectable in 30-40% of patients [2], typically associated with specific autoantibodies directed against myeloperoxidase (MPO). With no validated diagnostic criteria, heterogenous clinical presentations and strong overlap with hypereosinophilic syndrome (HES), the diagnosis can be difficult, however there are a number of classification criteria with established sensitivity and specificity that can be adapted to clinical practice [3]. Glucocorticoids are the mainstay of remission induction in all patients, with the addition of either cyclophosphamide or rituximab in patients with a Five
Factor Score of ≥1 [3]. For those with mild relapsing or refractory EGPA already established on glucocorticoids, the anti-interleukin-5 (IL-5) monoclonal antibody, mepolizumab improved rates of remission and allowed reduced glucocorticoid use compared to placebo, however remission was not obtained in 47% of patients [4]. The anti-IL5α receptor antibody, benralizumab, was shown to be non-inferior to mepolizumab in a randomized controlled trial of 140 patients with EGPA [5]. Regardless of these advances, glucocorticoid-related toxicity remains a major issue as only a small percentage of patients are able to be weaned off glucocorticoids despite remission maintenance therapy with rituximab, mepolizumab, benralizumab or conventional synthetic disease modifying anti-rheumatic drugs (csDMARDs) [3].
Janus kinase inhibitors (JAKi) provide a mechanistically sound theoretical option for the treatment of EGPA, with potential relevance both in remission-induction and as a glucocorticoid-sparing therapy.
Janus kinases are known to regulate the downstream signalling of various cytokines
involved in eosinophil regulation and migration, including IL-5 (via the JAK2/STAT5 axis) and
IL-4 (via the JAK1-3/STAT3 – 6 axis) [6-10]. Considering the well described role of these cytokines in the pathogenesis of EGPA, a biological rationale exists for a potential therapeutic benefit effect of inhibiting JAK1, 2 and/or 3 in the management of EGPA and of other eosinophilic disorders.
Clinical evidence on this topic is limited. The first case series reporting the potential for JAKi therapy in EGPA was recently published by Liu et al [11]. In this study, the authors described 11 patients with EGPA treated with tofacitinib 5mg bid, achieving a response rate of 100% with complete remission in 60% at 6 months and 80% at 9 months. After commencing tofacitinib, patients experienced normalisation of both ESR and CRP as well as significant reductions in both peripheral eosinophil counts and glucocorticoid doses. Beyond this study are reports of individuals with eosinophilic disorders, including one patient with drug reaction with eosinophilia and systemic symptoms (DRESS) [12] and thirteen patients with hypereosinophilic syndrome (HES) [13-16]. Here, we report our collective experience of 5 cases of EGPA who received JAKi therapy.
Methods
Physicians with an interest in vasculitis were contacted in 2024 and early 2025 in order to identify patients with EGPA (meeting the American College of Rheumatology / European Alliance of Associations for Rheumatology (ACR/EULAR) classification criteria [17] or the MIRRA trial criteria [4]) who had received treatment with a JAKi (tofacitinib, baricitinib, upadacitinib or ruxolitinib). Physicians were contacted through the following organizations: the Asia-Pacific League of Associations for Rheumatology Vasculitis Special Interest Group, the Australia and New Zealand Vasculitis Society, the Canadian Vasculitis Research Network, the European EGPA Study Group, and the Vasculitis Clinical Research Consortium.
Results
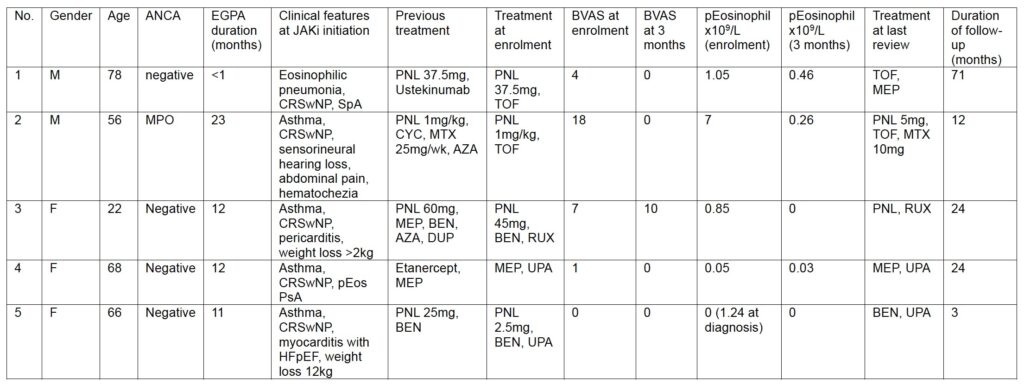
Five patients were identified, two from Australia (managed in private practice), one from Canada (tertiary referral centre), one from France (tertiary referral centre) and one from Italy (tertiary referral centre). All participants provided written, informed consent.
Case 1:
A 78 year old man with HLA B-27 negative peripheral and radiographic axial spondyloarthritis, treated with ustekinumab presented in 2020 with eosinophilic pneumonia with a peripheral eosinophil count of 1.05 x109/L, requiring 37.5mg prednisolone. His history included a 40-year history of chronic rhinosinusitis with polyposis (CRSwNP), asthma and two prior episodes of antibiotic-resistant and steroid-responsive episodes of pneumonia. A diagnosis of ANCA-negative EGPA was made. As it was not immediately possible to access mepolizumab, tofacitinib 5mg bid was commenced given its specificity for both JAK1 and JAK3 in the context of emerging literature supported targeting JAK3 in managing eosinophilic disease [12,18].
A dramatic clinical improvement was noted within seven days of commencing tofacitinib, accompanied by normalisation of the peripheral eosinophil count, allowing cessation of the prednisolone and inhaled corticosteroids within a fortnight.
Subsequently, due to concerns about cardiovascular risk [19], tofacitinib was stopped for two weeks. This precipitated acute sinusitis with epistaxis and acute dyspnoea, pain at the soles of the feet, blurred vision and a widespread pustular eruption; the peripheral eosinophil count again rose to 1.05×109/L, despite prednisolone 25mg/d. Within 24 hours of resuming tofacitinib, dyspnoea, rash and the blurred vision had resolved. Despite cessation of the prednisolone, the peripheral eosinophil count was 0.00 ten days later. The sinus congestion persisted; CT demonstrated nasal polyps that were subsequently resected.
Clinical remission has been maintained whilst on regular tofacitinib. Whenever tofacitinib has been withheld, the patient’s symptoms have flared, requiring multiple courses of prednisolone. Following the addition of mepolizumab to the treatment regimen, the severity of the flares experienced whenever tofacitinib has been withheld have diminished but have not resolved.
Case 2:
A 56-year-old man with a history of adult-onset asthma and CRSwNP presented with acute right-sided sensorineural hearing loss, abdominal pain, hematochezia, peripheral eosinophilia (absolute count: 7 × 10⁹/L), eosinophils on bronchoalveolar lavage, and a C-reactive protein (CRP) of 187 mg/L. Chest CT was unremarkable. MPO-ANCA was positive, while the remainder of his autoimmune serology was negative. A diagnosis of EGPA was made. Renal, cardiac, and cutaneous assessments were normal.
He was treated with high-dose glucocorticoids and intravenous cyclophosphamide for three months, followed by subcutaneous methotrexate at 25 mg weekly as maintenance. Clinical remission was achieved and sustained for two years, at which time, he developed constitutional symptoms, recurrent abdominal pain, hematochezia, marked peripheral eosinophilia, and elevated inflammatory markers. Audiometry confirmed new left-sided sensorineural hearing loss. Colonoscopy revealed features suggestive of inflammatory bowel disease, but biopsies were non-specific, and a definitive diagnosis was not established.
He was started on oral prednisone at 1 mg/kg, with clinical improvement. Azathioprine was trialled but discontinued due to gastrointestinal intolerance. Given the concern for EGPA-associated gastrointestinal vasculitis and possible inflammatory bowel disease overlap, tofacitinib was initiated at 5 mg orally twice daily to target both eosinophilic and lymphocytic inflammatory pathways. Subcutaneous methotrexate was continued at a reduced dose of 10 mg weekly. Over the subsequent 12 months, the patient achieved and maintained clinical remission on tofacitinib, methotrexate, and prednisone tapered to 5 mg daily. His gastrointestinal symptoms resolved completely, and no further ENT or respiratory flares occurred.
Case 3:
A 22-year old female was referred for EGPA and refractory severe asthma in 2020. She was a non-smoker, and had a history of CRSwNP since 2015, peripheral blood eosinophilia since 2016 (1.4 × 10⁹/L), asthma since 2017, and acute pericarditis in 2018. In 2020, she developed severe asthma with persistent airflow obstruction requiring oral glucocorticoids, palpable purpura, myalgias, weight loss, eosinophilic pneumonia and hypereosinophilic bronchiolitis. ANCA-negative EGPA was diagnosed. Initiation of mepolizumab was initially followed by a dramatic clinical response, allowing tapering of prednisone from 60 to 10 mg/d, and improvement of lung function. In 2021, asthma worsened, with severe airflow obstruction, and peripheral eosinophils were 0.7 × 10⁹/L while receiving 20 mg/d of prednisone. Investigations for chronic eosinophilic leukemia and secondary causes of eosinophilia were negative. Changes in management were unsuccessful, including an increase in mepolizumab dose (300 mg Q4W), switch to benralizumab (30 mg Q4W), azathioprine, dupilumab, and azithromycin, together with high-dose inhaled corticosteroids. The peripheral eosinophil count was 0.85 × 10⁹/L while receiving 45 mg/d of prednisone. Initiation of ruxolitinib as proposed by a nation-wide multidisciplinary discussion was followed by a dramatic clinical improvement, and increase in FEV1 from 36% to 76% of predicted values while prednisone was tapered to 30 mg/d. Unfortunately, the patient was later hospitalized in the ICU for acute severe asthma, severe abdominal pain and weight loss. She was discharged after glucocorticoids were increased; ruxolitinib was continued.
Case 4:
A 68 year old female receiving etanercept for psoriatic arthritis presented in 2022 with asthma, rhinosinusitis with nasal polyposis and a peripheral eosinophilia. EGPA was diagnosed and mepolizumab commenced. In 2023 her inflammatory arthritis flared, however her EGPA remained under good control. Her inflammatory arthritis responded significantly to a change from etanercept to upadacitinib 15mg daily; the clinical severity of her EGPA did not change. No adverse safety signals were noted.
Case 5:
A 66 year old female with a 15 year history of CRSwNP developed rapidly progressive adult onset asthma and months later was admitted in August 2024 with severe eosinophilic asthma, heart failure with preserved ejection fraction (HFpEF) and myocarditis on MRI. Prednisolone 25mg and benralizumab 30mg sc monthly were commenced, along with pharmacological management of her HFpEF. Her peripheral eosinophilia normalised and by the time of her presentation to the rheumatology clinic in February 2025 (when the diagnosis of EGPA was made) had weaned off prednisolone entirely. At that time she reported only mild dyspnoea after 20 minutes walking on the flat. An endomyocardial biopsy performed 7 months after the commencement of benralizumab was not diagnostic. In April 2025 she represented with steroid responsive dyspnoea and heart rate variability, with recrudescence of her symptoms after the prednisolone was weaned from 25mg to 2.5mg. Upadacitinib 15mg daily was commenced and the prednisolone was ceased two weeks later. Upadacitinib was withheld perioperatively for excision of a nasiolabal squamous cell carcinoma, during which time her dyspnoea and heart rate variability returned. Both slowly returned to baseline over three weeks after upadacitinib was recommenced.
Discussion
These five cases provide further depth to our understanding of the potential for JAKi therapy in the management of EGPA. Several sets of classification criteria have been developed for the diagnosis of EGPA, however none have been validated and there are currently no diagnostic criteria [3]. All five patients described in this paper met at least one of the accepted classification criteria for EGPA. In contrast, it was not clear from the clinical descriptions provided that most of the patients described by Liu et al actually had EGPA [11]. Although they had eosinophilic disease, only three of the 11 patients were reported to have
asthma and only four were reported to have sinusitis. The presence or absence of sinonasal polyposis was not described. Three patients were described to have peripheral nerve involvement. Five of the 11 patients had no listed history of asthma, sinusitis or peripheral neuropathy of any type (patients 1, 4, 7, 8, 11). Therefore we may conclude from the paper by Liu et al that tofacitinib may be effective in eosinophilic diseases, however it is difficult to draw a strong conclusion about its efficacy specifically in EGPA. Asthma, sinonasal polyps (not all types of sinusitis) and mononeuritis multiplex are the three fundamental clinical characteristics of EGPA. The two patients described in our paper who received tofacitinib appeared to have the most dramatic clinical responses, though it is difficult to draw strong conclusions from such a small sample size, particularly when two of the patients described (both of whom received upadacitinib) did not have objective evidence of clinically significant EGPA activity at the time JAKi therapy was commenced. Nevertheless, the impressive clinical responses to tofacitinib and the fact that patient #3 required admission to ICU for severe asthma whilst on ruxolitinib does raise the question as to whether there is a clinical advantage in targeting JAK3 with tofacitinib. In spite of these issues, the cases presented both in this paper and in the article by Liu et al provide a strong argument for a formal randomized controlled trial comparing JAKi therapy to the current standard of care for EGPA.
References:
- Churg, Strauss. Am J Pathol 1951; 27(2):277-301
- Moiseev S, Bossuyt X, Arimura Y, et al. International Consensus on ANCA Testing in Eosinophilic Granulomatosis with Polyangiitis. Am J Respir Crit Care Med 2020; Jun 25. doi: 10.1164/rccm.202005-1628SO
- Emmi G, Bettiol A, Gelain E, et al. Evidence-Based Guideline for the diagnosis and management of eosinophilic granulomatosis with polyangiitis. Nat Rev Rheumatol 2023; 19(6):378-393. doi: 10.1038/s41584-023-00958-w
- Wechsler ME, Akuthota P, Jayne D, et al. Mepolizumab or Placebo for Eosinophilic Granulomatosis with Polyangiitis. N Engl J Med 2017; 376(20):1921-32. doi: 10.1056/NEJMoa1702079
- Wechsler ME, Nair P, Terrier B, et al. Benralizumab versus Mepolizumab for Eosinophilic Granulomatosis with Polyangiitis. N Engl J Med 2024; 390(10):911-921. doi: 10.1056/NEJMoa2311155
- Huang I-H, Chung W-H, Wu P-C, Chen C-B. JAK-STAT signaling pathway in the pathogenesis of atopic dermatitis: An updated review. Front Immunol 2022; 13:1068260. doi:10.3389/fimmu.2022.1068260
- Dubois GR, Schweizer RC, Versluis C, et al. Human eosinophils constitutively express a functional interleukin-4 receptor: interleukin-4 -induced priming of chemotactic responses and induction of PI-3 kinase activity. Am J Respir Cell Mol Biol 1998; 19(4):691 9. doi:10.1165/ajrcmb.19.4.3208
- Junttila IS. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front Immunol 2018; 9:888. doi:10.3389/fimmu.2018.00888
- Nonaka M. Nonaka R, Woolley K, et al. Distinct immunohistochemical localization of IL-4 in human inflamed airway tissues. IL-4 is localized to eosinophils in vivo and is released by peripheral blood eosinophils. J Immunol 1995; 155(6):3234. https://doi.org/10.4049/jimmunol.155.6.3234
- Stritesky GL, Muthukrishnan R, Sehra S, et al. The transcription factor STAT3 is required for T helper 2 cell development. Immunity 2011; 34(1):39 49. doi: 10.1016/j.immuni.2010.12.013
- Liu Y, Zhan M, Chen R et al. Tofacitinib in combination with glucocorticoids in the treatment of eosinophilic granulomatosis with polyangiitis: a pilot study of 11 cases. Rheumatology (Oxford) 2025; Aug 18:keaf397. doi: 10.1093/rheumatology/keaf397
- Kim D, Kobayashi T, Voisin B, et al. Targeted therapy guided by single-cell transcriptomic analysis in drug-induced hypersensitivity syndrome: a case report. Nat Med 2020; 26(2):236 243. doi: 10.1038/s41591 019 0733 7
- Walker S, Wang C, Walradt T, et al. Identification of a gain-of-function STAT3 mutation (p.Y640F) in lymphocytic variant hypereosinophilic syndrome. Blood 2016; 127(7):948 51. doi: 10.1182/blood 2015 06 654277
- King B, Lee AI, Choi J. Treatment of Hypereosinophilic Syndrome with Cutaneous Involvement with the JAK Inhibitors Tofacitinib and Ruxolitinib. J Invest Dermatol 2017; 137(4):951 954. doi: 10.1016/j.jid.2016.10.044
- Šteňová E, Tarabčáková L, Babál P. et al. Hypereosinophilic syndrome-a rare adverse event of anti-cytokine treatment in rheumatoid arthritis resolved after Janus kinase inhibitor therapy. Clin Rheumatol 2020; 39(11):3507 3510. doi: 10.1007/s10067 020 05134
- Faguer S, Groh M, Vergez F, et al. JAK inhibition for CD3- CD4+ lymphocytic-variant hypereosinophilic syndrome. Clin Immunol 2023; 251:109275. doi: 10.1016/j.clim.2023.109275
- Grayson P, Ponte C, Suppiah R, et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Eosinophilic Granulomatosis with Polyangiitis. Ann Rheum Dis 2022; 81(3):309-314. doi: 10.1136/annrheumdis-2021-221794
- Kudlacz E, Conklyn M, Andresen C, et al. The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur J Pharmacol 2008; 582(1-3):154-61. doi: 10.1016/j.ejphar.2007.12.024
- Ytterberg S, Bhatt D, Mikuls T et al. Cardiovascular and Cancer Risk with Tofacitinib in Rheumatoid Arthritis. N Engl J Med 2022; 386:316-326 DOI: 10.1056/NEJMoa2109927